Molecular Modeling and In Silico Evaluation of Novel Pyridazinones Derivatives as Anticonvulsant Agents
Husain A, Khokra SL, Thakur P, Choudhary D, Kohli S, Ahmad A and Khan SA
DOI10.21767/2469-6692.10007
1Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Jamia Hamdard (Hamdard University), New Delhi-110064, India.
2Institute of Pharmaceutical Sciences, Kurukshetra University, Haryana-136119, India.
3Health Information Technology Department, Jeddah Community College, King Abdulaziz University, Jeddah, Saudi Arabia.
4Department of Pharmacy, Oman Medical College, Muscat, Sultanate of Oman.
- *Corresponding Author:
- Asif Husain
Department of Pharmaceutical Chemistry
Faculty of Pharmacy, Jamia Hamdard (Hamdard University)
New Delhi-110064
E-mail: asifhusain@yahoo.com
Abstract
Context: Virtual screening techniques or computational methods are used for the drug discovery and development.
Objective: In search for the safer and effective anticonvulsant agents, docking study on pyridazinone derivatives was performed to potentiate GABA mediated chlorine channel opening.
Methods: A total of eighteen compounds were screened for their anticonvulsant activity using molecular docking inside the ligand binding domain of PDB ID 2Q1Q using Molegro Virtual docker. QikProp 3.4 &
molinspiration software were also used to evaluate drug likeliness and to perform comparative bioactivity analysis for all the pyridazinone derivatives.
Results: Out of eighteen compounds, (I) showed highest Mol dock score and (XVIII) formed maximum number of hydrogen bond interactions. Oxygen atom of the Nitro group on the biphenyl ring of compound (XV) formed a strong hydrogen bond with Thr199 with a distance 2.33?. Mol dock score of diazepam (standard drug) was -101.5306.
Conclusions: Based on the results of mol dock score and number of hydrogen bond interactions, compounds (XVIII) and (XIII) observed to be the most potent compounds.
Keywords
Convulsant; ADME; Docking; Heterocyclic; Pyridazinone.
Introduction
The term ‘In silico’ or computational methods are virtual screening techniques used for the discovery of novel molecules of potential pharmaceutical interests and for advancement in therapeutics. The in silico studies considered as complementary to in vivo and in vitro biological studies are performed by using a computer and are playing increasingly larger and more important role in drug discovery and development. Computational methods include, docking, homology modeling, quantitative structure activity relationships (QSARs), similarity and pharmacophore searching, virtual ligand screening, data mining, and data analysis tools [1]. These tools are becoming increasingly popular in drug design and the last decade has witnessed an upsurge in the development and application of computational methods applied to the drug development [2]. These methods are used in the drug discovery and optimization of novel molecules having potential affinity and specificity for the selected therapeutic targets. The pharmacokinetic (PK) properties of a drug molecule such as Absorption, Distribution, Metabolism, Excretion & toxicity (ADME-T) depend more or less on the chemical descriptors of molecule and have become quite important in the drug-discovery process. Prediction and estimation of PK properties by using computational methods have become increasingly important in drug selection, promotion process and also are promising tools for the fast screening of potential drug candidate [3]. Epilepsy is a chronic neurological disorder that is characterized by recurrent unprovoked seizures [4]. Traditionally, pharmacological strategies for the treatment of epilepsy are aimed at suppressing the initiation or propagation of seizures rather than the treatment of the underling processes that lead to epilepsy [5]. Because of this approach, the epilepsy is usually controlled but not cured with the anticonvulsants drugs. The correct approach in the treatment of epilepsy should be to identify new targets through better understanding of molecular mechanism of epilepsy in first place. Second strategy should aim at modification of already existing drugs and formulations [6]. Anticonvulsant agents act by more than one mechanism of action which include prolong inactivation of the ion channel or potentiating neurotransmitter system [7- 9]. In fact some of the clinically used anticonvulsant drugs have not been linked with the specific site in the brain, and their exact mechanism of action remain unknown [10]. In the past, conventional screening and/or structural modification methods were used for the discovery of novel anticonvulsant agents rather than a mechanism-driven drug design process. Therefore, drug identification was generally carried out on the basis of seizures type i.e. in vivo screening tests rather than etiology of the disease. In recent years, considerable emphasis has been placed on the relation between epileptic seizures and neurotransmitter in the brain. Augmenting the inhibition of neurotransmission or diminishing the excitement of transmission provides a great approach to the design of anticonvulsant and anti-epileptic drugs. γ-Aminobutyric acid (GABA) is a good example of inhibitory neurotransmission. GABA being water soluble is difficult to pass through the blood-brain barrier (BBB) and thus penetrates poorly into the brain; therefore, it cannot be used as an anti-epileptic in clinics. Hence, to improve its lipid solubility or ability to cross the BBB, a series of arylpyridazine derivatives of GABA were designed [11,12]. Pyridazinones are the derivatives of pyridazine possessing a magic azomethine (-NH-N=CH-) moiety. The pyridazinone are six membered heterocyclic compounds that also contains a cyclic amide moiety in their ring structure that plays an important role in exhibiting various pharmacological activity ranging from cardiovascular properties, anti-inflammatory, antidiabetic, analgesic, anti-AIDS, anti-cancer, anti-microbial and anticonvulsant activities [13,14]. Thus pyridazinone appear to be potential drug candidates for the anticonvulsant therapy. In order to develop potent and safe anticonvulsant drugs, many researchers have synthesized the different pyridazine and pyridazinone derivatives and evaluated for their anticonvulsant activity in different animal models of seizures [15]. They observed that presence of a phenyl substitution on pyridazinone ring with electron withdrawing group possess good activity against tonicclonic seizures. Using this background information and in line of our current interest in the protein ligand binding, we describe herein the in silico study of various hypothetical pyridazinones and their interaction with facilitator of GABA transmission by computational docking method. The compounds found to have high Mole Dock Score and formed maximum number of H-bond interaction were synthesized and evaluated for their anticonvulsant activities. However computational data based on docking studies is presented in this research paper.
Materials and methods
Docking is the computational method, which predicts the preferred orientation of one molecule to a second, when bound to each other to form a stable complex. Docking study was performed with a set of hypothetical pyridazinone derivatives using Molegro Virtual docker MVD 2010.4.2 on X-ray structures of GABA (PDB ID: 2Q1Q) downloaded from the protein data bank (PDB).
Ligand preparation
The molecules were draen in MarvinSketch 5.11.4. After converting the 2D molecules into 3D, the conformational energy of molecules was minimized. The resulting structures were saved in MarvinSketch as MDL Molfile (*.mol). The simplest use of MarvinSketch produces a single, low energy with 3D structure. Finally, the prepared 3D structures of molecules were imported by dragging or dropping a molecule structure file in the workspace.
Protein preparation and detecting cavities of protein molecules
The GABA facilitator (PDB ID 2QIQ) X-ray structures were accessed from the protein data bank (PDB) and imported. The protein structures were prepared using the protein preparation wizard in MVD. In this step, bond order were assigned, all hydrogen’s in the structure were added, and bonds to metals were deleted and adjust the formal charge on the metal and the neighbouring atoms and deleted waters that were more than 5Å specific distance. The energy of imported molecules was minimized using Ligand Energy inspector. The energy minimization supports in stability of molecules to be imported. Next, the protein surface was created using protein preparation. This step helps to inspect and change the protonation state for the residues. In order to determine the potential binding sites, a cavity prediction algorithm was performed.
Executing a docking set up through docking wizard panel
Molegro searches for suitable interaction between one or more ligand molecules and the receptor. Further, all ligands were selected and docking was performed through the docking wizard panel. After this step docking results were imported. Various orientation (or poses) of docked compounds were analysed by determination of Mol Dock Score and H-bond interaction. Compounds were arranged according to their Mol Dock scores and were visualized inside the pocket to view their fitting and closure to main residues. Molecular docking studies revealed further insight into the nature of interactions between the compounds and the active site amino acids to rationalize the obtained biological results. By this docking study we came to know that most of our designed ligands are interacting to GABA facilitated target with sufficient selectively and specificity. The docking analysis is done and the results are presented in the form of image given in (Figure 1-3), furthermore MolDock score and hydrogen bond interactions between receptor and ligands are presented in the Table 1 (see supplementary material).
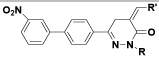 |
||
| Compound no. | R | R’ |
| (I) | C6H5 |  |
| (II) | C6H5 | 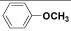 |
| (III) | C6H5 |  |
| (IV) | H | 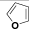 |
| (V) | H | 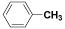 |
| (VI) | H |  |
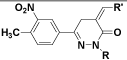 |
||
| (VII) | C6H5 | 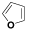 |
| (VIII) | C6H5 | 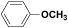 |
| (IX) | C6H5 |  |
| (X) | H | 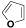 |
| (XI) | H |  |
| (XII) | H |  |
| (XIII) | H |  |
| (XIV) | H |  |
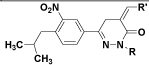 |
||
| (XV) | H | 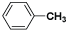 |
| (XVI) | H | 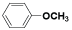 |
| (XVII) | H | 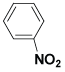 |
| (XVIII) | H | 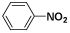 |
Diazepam (Standard drug) |
||
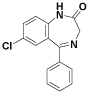
Table 1: Structures of all the hypothetical pyridazinone derivatives & standard drug.

Figure 1: Binding affinity of synthesized compound in specific cavity.

Figure 2: Superimposition of synthesized compounds with internal ligand in active site.

Figure 3: Docking images of pyridazinone derivatives (I-XVIII) showing hydrogen bonds with 2QIQ protein residues.
Ligand based ADME&Toxicity studies
ADME-T properties were calculated using Qikprop 3.6 tool of Schrodinger 2014 & molinspiration. All the structures showed significant values for the properties analyzed. Tables 2-5, additionally showed drug-like characteristics based on Lipinski’s rule of 5 [16]. Qikprop predicts physically significant descriptors and pharmaceutically relevant properties like absorption, distribution, metabolism, excretion and toxicity (ADMET) & acceptability of compounds based on Lipinski’s rule of five, which is essential to ensure drug-like pharmacokinetic profile while using rational drug design & Molinspiration, an online tool, used to perform QSAR studies in order to identify potential activators of biological targets.
| Compound | Mol Dock Score | No. of H-bond | H-bond distance Angstrom |
Amino acid involved | Structural feature |
|---|---|---|---|---|---|
| Diazepam (Standard drug) |
-101.530 | 4 | 3.00 3.48 3.54 3.21 |
Thr199 Thr199 Thr200 Thr200 |
O – O N – O N – N N - O |
| (I) | -135.22 | 3 | 3.36 3.39 3.35 |
Gln92 Thr200 Gly132 |
N of pyridazinone with N O of ketone with O O of NO2with O |
| (II) | -96.6903 | 6 | 3.13 3.10 3.21 3.26 2.60 3.58 |
Gln92 Phe95 Phe66 Asn244 Asn244 Phe95 |
N of pyridazinone with N O of NO2 with N O of NO2 with N N of NO2 with N O of NO2 with N N of NO2 with N |
| (III) | -129.905 | 5 | 3.09 3.57 2.92 |
Gln92 Gln92 Gln92 Gly132 Gly132 |
N of pyridazinone with N N of pyridazinone with N N of pyridazinone with N O of NO2 with N O of NO2 with N |
| (IV) | -105.079 | 3 | 3.47 3.03 3.38 |
Tyr7 Thr200 Thr199 |
O of furfuryl with O N of pyridazinone with O O of NO2 with O |
| (V) | -106.233 | 4 | 3.10 2.83 3.25 3.18 |
Phe131 Gly132 Gly132 Thr200 |
O of NO2 with N O of NO2 with N N of NO2 with N O of ketone with O |
| (VI) | -100.213 | 3 | 3.04 3.09 3.15 |
Asn244 His96 Thr200 |
O of OCH3 with N O of OCH3 with N N of pyridazinone with O |
| (VII) | -125.146 | 6 | 2.60 3.17 3.11 2.90 3.18 3.41 |
Trp5 Trp5 Thr199 Thr199 Thr199 Thr199 |
O of NO2 with N N of NO2 with N O of furfuryl with O O of furfuryl with N N of pyridazinone with O N of pyridazinone with O |
| (VIII) | -124.121 | 5 | 2.58 3.47 2.74 3.04 2.55 |
His96 His96 Tyr7 Tyr7 Tyr7 |
O of NO2 with N N of NO2 with N O of NO2 with O N of NO2 with O O of NO2 with O |
| (IX) | -118.998 | 6 | 2.73 3.15 2.71 3.37 2.59 3.34 |
Asp62 His119 Thr199 Thr199 Thr199 Thr199 |
O of ketone with N O of ketonewith N O of NO2 with O N of NO2 with N O of NO2 with N N of NO2 with O |
| (X) | -115.379 | 9 | 2.90 3.07 3.10 2.77 3.10 2.60 2.87 2.36 3.04 |
Asn62 Asn67 Gln92 Thr199 Thr199 Thr199 Thr200 Thr200 Thr200 |
O of ketone with N N of pyridazinone with O N of pyridazinone with N O of NO2 with O N of NO2 with O O of NO2 with O O of NO2 with N O of NO2 with N N of NO2 with N |
| (XI) | 100.627 | 8 | 2.70 3.40 3.13 2.98 2.59 3.05 3.21 2.60 |
Asn62 Asn67 Gln92 His119 Thr199 The199 Thr199 Thr199 |
O of ketone with N N of pyridazinone with O N of pyridazinone with N O of NO2 with N O of NO2 with O N of NO2 with O N of NO2 with N O of NO2 with N |
| (XII) | -115.777 | 8 | 2.64 3.44 3.11 3.05 2.60 3.10 3.34 2.54 |
Asn62 Asn67 Gln62 His119 Thr199 Thr199 Thr199 Thr199 |
O of ketone with N N of pyridazinone with O N of pyridazinone with N O of NO2 with N O of NO2 with O N of NO2 with O N of NO2 with N O of NO2 with N |
| (XIII) | -123.657 | 5 | 3.33 3.45 3.36 2.43 3.29 |
Asn244 His96 Tyr7 Tyr7 Gln92 |
O of NO2 with N N of NO2 with N N of NO2 with O O of NO2 with O N of pyridazinone with O |
| (XIV) | -127.393 | 6 | 3.10 3.56 2.98 2.60 3.11 2.20 |
Trp5 His4 Thr199 Thr199 Thr199 Thr199 |
N of NO2 with N O of NO2 with N N of NO2 with O O of NO2 with N N of NO2 with N O of NO2 with O |
| (XV) | -101.083 | 10 | 2.95 3.01 3.12 3.38 2.60 3.22 2.33 3.01 2.97 2.55 |
Asn62 Asn62 Gln92 His119 Thr199 Thr199 Thr199 Thr200 Thr199 Thr199 |
O of ketone with N N of pyridazinone with O N of pyridazinone with N O of NO2 with N O of NO2 with O N of NO2 with N O of NO2 with N O of NO2 with N N of NO2 with O O of NO2 with O |
| (XVI) | -108.17 | 6 | 3.41 2.85 2.41 3.10 2.95 2.18 |
Gln92 Asn62 Asn62 Asn67 Asn67 Asn67 |
N of pyridazinone with N N of NO2 with N O of NO2 with N N of NO2 with N O of NO2 with N O of NO2 with N |
| (XVII) | -118.136 | 11 | 2.96 3.04 3.50 2.89 3.31 3.32 2.76 2.66 2.77 3.14 3.09 |
Asn62 Asn67 His64 Trp5 Gln92 Thr200 Thr200 Thr200 Thr200 Thr200 Thr200 |
O of ketone with N N of NO2 with O O of NO2 with N N of NO2 with N N of NO2 with N O of NO2 with O O of NO2 with N O of NO2 with O O of NO2 with O N of NO2 with O N of NO2 with N |
| (XVIII) | -104.514 | 6 | 3.28 3.10 2.89 2.88 2.38 2.28 |
Gln92 Asn67 Asn62 Ala65 Asn62 Asn67 |
N of pyridazinone with N N of NO2with N N of NO2 with N O of NO2 with N O of NO2 with N O of NO2 with N |
Table 2: Mol dock score and bonding interaction of pyridazinone derivatives with amino acids resides using PDB ID: 2Q1Q for determination of anticonvulsant activity.
| Compounds | # star | #rotor | MW | QPLog Khsa | SASA | Volume | Donor HB | Accpt HB | QP log P OCT | QP log PW |
| (I) | 4 | 4 | 489.529 | 0.876 | 844.932 | 1518.065 | 0.000 | 3.750 | 22.293 | 9.149 |
| (II) | 1 | 5 | 397.432 | 1.156 | 723.075 | 1264.316 | 0.000 | 3.000 | 19.879 | 9.001 |
| (III) | 4 | 4 | 473.530 | 1.311 | 839.435 | 1502.411 | 0.000 | 3.000 | 21.967 | 8.611 |
| (IV) | 0 | 4 | 373.367 | 1.540 | 659.454 | 1143.330 | 1.000 | 3.500 | 18.694 | 9.512 |
| (V) | 4 | 4 | 473.530 | 2.007 | 839.435 | 1502.411 | 0.000 | 3.000 | 21.967 | 8.611 |
| (IV) | 1 | 5 | 413.432 | 1.810 | 728.168 | 1279.570 | 1.000 | 3.750 | 20.202 | 9.536 |
| (VI) | 1 | 3 | 387.394 | 0.434 | 688.186 | 1207.864 | 0.000 | 3.500 | 17.773 | 8.016 |
| (VIII) | 1 | 4 | 427.459 | 0.710 | 751.932 | 1338.347 | 0.000 | 3.750 | 19.250 | 7.994 |
| (IX) | 2 | 3 | 411.459 | 0.859 | 746.411 | 1322.665 | 0.000 | 3.000 | 18.924 | 7.456 |
| (X) | 0 | 3 | 311.296 | 1.061 | 566.105 | 963.355 | 1.000 | 3.500 | 15.652 | 8.356 |
| (XI) | 0 | 3 | 335.362 | 1.528 | 630.336 | 1084.930 | 1.000 | 3.000 | 16.843 | 7.848 |
| (XII) | 0 | 4 | 351.361 | 1.330 | 635.473 | 1100.221 | 1.000 | 3.750 | 17.165 | 8.838 |
| (XIII) | 0 | 4 | 380.359 | 0.753 | 668.161 | 1162.677 | 0.000 | 4.000 | 17.724 | 7.526 |
| (XIV) | 0 | 4 | 380.359 | 0.752 | 668.042 | 1162.301 | 0.000 | 4.000 | 17.719 | 7.525 |
| (XV) | 1 | 5 | 377.442 | 1.213 | 703.281 | 1243.522 | 1.000 | 3.000 | 18.332 | 7.409 |
| (XVI) | 1 | 6 | 408.413 | 1.001 | 709.572 | 1256.606 | 1.000 | 4.000 | 20.273 | 8.832 |
| (XVII) | 1 | 6 | 408.413 | 1.000 | 709.444 | 1256.214 | 1.000 | 4.000 | 20.268 | 8.831 |
| (XVIII) | 1 | 6 | 408.413 | 1.001 | 709.572 | 1256.606 | 1.000 | 4.000 | 20.273 | 8.832 |
Table 3: Results of ligand based ADME-T studies of proposed pyridazinone derivatives.
| Compounds | QP log PO/W | QP Log S |
QP LogBB | QPPCaco | QPPMDCK | QPLog Kp | HUMAN ORAL ABSORPTION % | PSA | RULE OF 5 | HOA |
| (I) | 6.463 | -8.537 | -1.126 | 171.153 | 281.924 | -1.371 | 100.000 | 86.673 | 1 | 1 |
| (II) | 7.168 | -9.451 | -1.226 | 188.973 | 290.690 | -1.351 | 100.000 | 87.296 | 1 | 1 |
| (III) | 7.430 | -9.902 | -1.167 | 188.912 | 290.492 | -1.451 | 100.000 | 79.008 | 1 | 1 |
| (IV) | 4.200 | -6.359 | -1.605 | 594.370 | 73.406 | -3.053 | 91.512 | 104.190 | 0 | 1 |
| (V) | 5.189 | -7.711 | -1.654 | 611.063 | 81.672 | -3.043 | 85.108 | 96.495 | 1 | 1 |
| (VI) | 4.968 | -7.352 | -1.711 | 611.447 | 81.700 | -2.944 | 96.778 | 104.779 | 0 | 1 |
| (VII) | 5.173 | -6.937 | -1.306 | 212.618 | 92.804 | -1.843 | 95.634 | 85.163 | 0 | 1 |
| (VIII) | 5.878 | -7.852 | -0.903 | 234.548 | 103.192 | -1.823 | 100.000 | 85.786 | 1 | 1 |
| (IX) | 6.140 | -8.302 | -0.864 | 234.480 | 103.160 | -1.923 | 100.000 | 77.496 | 1 | 1 |
| (X) | 2.947 | -4.840 | -1.415 | 740.353 | 357.461 | -3.528 | 85.860 | 102.697 | 0 | 3 |
| (XI) | 3.927 | -6.177 | -2.155 | 761.108 | 368.305 | -3.517 | 92.362 | 95.004 | 0 | 3 |
| (XII) | 3.717 | -5.833 | -3.359 | 761.557 | 368.540 | -3.418 | 91.111 | 103.287 | 0 | 3 |
| (XIII) | 3.697 | -6.133 | -0.962 | 82.052 | 33.160 | -4.328 | 82.849 | 125.726 | 0 | 3 |
| (XIV) | 3.692 | -6.131 | -1.963 | 81.681 | 33.000 | -4.333 | 82.790 | 125.715 | 0 | 3 |
| (XV) | 4.973 | -7.184 | -1.464 | 277.299 | 123.664 | -3.246 | 100.000 | 93.836 | 0 | 1 |
| (XVI) | 3.956 | -6.754 | -2.559 | 33.257 | 12.493 | -4.949 | 77.350 | 138.754 | 0 | 1 |
| (XVII) | 3.952 | -6.752 | -2.562 | 33.108 | 12.433 | -4.954 | 77.290 | 138.741 | 0 | 1 |
| (XVIII) | 3.956 | -6.754 | -2.559 | 33.257 | 12.493 | -4.949 | 77.350 | 138.754 | 0 | 1 |
Table 4: Results of ligand based ADME-T studies of proposed pyridazinone derivatives
| Compound no. | GPCR ligand | Ion Channel Modulator | Kinase inhibitor | Nuclear receptor ligand | Protease inhibitor | Enzyme inhibitor |
| (I) | -0.32 | -0.38 | -0.60 | -0.34 | -0.53 | -0.39 |
| (II) | -0.23 | -0.31 | -0.37 | -0.16 | -0.33 | -0.26 |
| (III) | -0.22 | -0.33 | -0.36 | -0.15 | -0.33 | -0.27 |
| (IV) | -0.41 | -0.43 | -0.80 | -0.57 | -0.57 | -0.57 |
| (V) | -0.29 | -0.29 | -0.50 | -0.32 | -0.32 | -0.22 |
| (VI) | -0.29 | -0.30 | -0.30 | -0.34 | -0.23 | -0.23 |
| (VI) | -0.36 | -0.36 | -0.76 | -0.50 | -0.61 | -0.52 |
| (VIII) | -0.24 | -0.23 | -0.47 | -0.26 | -0.26 | -0.36 |
| (IX) | -0.23 | -0.20 | -0.47 | -0.27 | -0.27 | -0.35 |
| (X) | -0.36 | -0.57 | -0.86 | -0.83 | -0.77 | -0.70 |
| (XI) | -0.23 | -0.35 | -0.50 | -0.52 | -0.40 | -0.46 |
| (XII) | -0.20 | -0.38 | -0.49 | -0.50 | -0.42 | -0.47 |
| (XIII) | -0.29 | -0.31 | -0.47 | -0.49 | -0.38 | -0.43 |
| (XIV) | -0.30 | -0.31 | -0.46 | -0.49 | -0.38 | -0.43 |
| (XV) | -0.34 | -0.35 | -0.73 | -0.36 | -0.33 | -0.31 |
| (XVI) | -0.21 | -0.30 | -0.50 | -0.35 | -0.26 | -0.36 |
| (XVII) | -0.20 | -0.24 | -0.47 | -0.35 | -0.23 | -0.33 |
| (XVIII) | -0.31 | -0.29 | -0.67 | -0.33 | -0.28 | -0.26 |
| Diazepam | -0.13 | -0.37 | -0.61 | -0.34 | -0.12 | -0.01 |
Table 5: Results of drug likeness properties viz; bioactivity score of proposed pyridazinone derivatives by molinspiration.
Results and Discussion
Docking studies revealed that the all the compounds had good potentiating power towards GABA mediated Chlorine channel opening. Docking studies on hypothetically designed compounds of pyridazinone derivatives showed good docking results. Molegro Virtual Docker allows the flexible docking of ligand into its site of action. In the analysis of docking results, we found that the hypothetically design compounds selected for docking studies were able to interact with the hydrophobic pocket of the protein efficiently and shows good anticonvulsant activity. Substitution on phenyl ring with electron withdrawing group also showed good results. Most important feature of docking is the logical interaction of the ligand with the putative-binding site of enzyme. The molecules were built in MarvinSketch 5.11.4. The simplest use of MarvinSketch produces a single, low energy with 3D structure. The energy minimization supports in stability of molecules to be imported. Each search was continued until the global energy minima were carried. The protein structures were prepared using the protein preparation wizard in MVD and optimization of hydrogen bond network was carried out. The favourable interaction between one or more ligand molecules and a receptor molecule was carried out by detecting cavity where ever required, to ensure that possible activities were not missed. The process of evaluating a particular pose was achieved by counting the number of suitable interactions that might be hydrogen, hydrophobic or electrostatic bonding.
Based on this analysis, the residues Tyr7, Asp62, Asn62, Ala65, Phe66, Asn67, Gln92, Phe95, His119, Phe131, Gly132, Thr199, Thr200, Asn244, Trp6, Glu7, Thr89, Leu78, Arg67, Arg69, Ser90 and Asp47 were predicted to interact with GABA receptor and to act as functional residues. Mol Dock Score of Standard Compound Diazepam was-101.5306. Diazepam formed four hydrogen bonds interactions with two amino acids which are Thr199& Thr200 and the distance of respective amino acid is 3.00 (Å), 3.48(Å), 3.54 (Å) and 3.21(Å). Out of eighteen compounds, Compound (I) exhibited relatively comparable binding affinity with highest Mol Dock Score of -135.22 comparable to standard compound. Compound (XVIII) form maximum hydrogen bond interactions i.e. eleven hydrogen bond with six amino acid which are Trp5, Asn62, Asn67, His64, Gln92 & Thr200 the distance of respective amino acid are 2.89(Å), 2.96(Å), 3.04(Å), 3.31(Å), 3.32(Å), 2.76(Å), 2.66(Å), 2.77(Å), 3.14(Å) & 3.09 (Å). Oxygen of the Nitro group on the biphenyl ring of compound (I) formed a strong hydrogen bond with Thr199 with a distance 2.33(Å). Some other docked molecule also showed good Mol dock Score and H-bond interactions viz., (VII), (VIII), (X), (XII), (XIII), (XIV), (XVII), and (XVIII). On the basis of docking studies most potent anticonvulsant compounds in our study were found to be (I) and (XVIII), because these showed highest Mol dock Score and form maximum number of hydrogen bond interactions (Table 2). The compounds that had highest Mol Dock Score and minimum energy can be used for further drug designing and also provides a way to synthesize new potent compounds in laboratory. Drug likeliness, stars, log P, log S, molecular weight, QPlogBB, percent human oral absorption, PSA, SASA etc. may be used to estimate the compound’s overall potential to achieve a ligand as potential drug candidate. The most important pharmaceutical descriptor in qikprop is stars, which are the combination of all of all properties. A few numbers of stars reflects more drug likeness in the range. Out of eighteen deigned compounds, six compounds showed 0 values, and eight compounds showed 1 value of stars. Range of some other important descriptor were, molecular weight of the designed compounds fall in between the range 311-650 Daltons, SASA range were 566.105-844.932, estimated number of HBdonor by the solute to water molecules in the aqueous solutions were 0.000-1.000, Estimated number of accept HB by the solute to water in aqueous solutions were 3.000-4.000, predicted QP log POCT were 15.652-22.355, predicted QP log PW were 7.525-8.440, predicted QP log PO/W were 2.947-7.430, predicted QP logS were -9.902-4.840, predicted QP logBB were-3.359-0.864, predicted human oral absorption were 82.790-100.000 and predicted PSA were 77.496-138.754. Out of eighteen deigned compounds, twelve compounds showed zero Lipinski violation and remaining compounds showed one Lipinski violation satisfying pharmacological properties of 95% available drugs high to medium estimated oral absorption availability. When the Lipinski violation is more than one at that time there is problem in oral bioavailability. The deigned compounds did not show toxic groups. All the analogs were neutralized before being used by Qikprop & molinspiration. All the structures showed significant values for the properties analyzed Table 3 and showed drug-like characteristics based on Lipinski’s rule of 5. The ligand based ADME-T values of design compound inhibitors are given in Table 4. The compounds obey Lipinski rule of five for molecular weight (mol MW) less than 500, solubility (QPlogS) greater than-4 number of ratable bond (#rotor) less than 10, and number of hydrogen bond donor and accepter (Hb-doner and accepter) are less than 5 and less than 10 respectively but show violations for partition coefficient between octanol and water (logP (O/W)) which is found between 2 to 6.5. Brain/blood partition coefficient (QPlogBB) parameter indicated the ability of the drug to pass through the blood brain barrier which is mandatory for enhancement of GABA-ergic inhibition. Whereas QPPMDCK predicted apparent MDCK cell permeability in nm/s. MDCK cells are considered to be a good mimic for the blood–brain barrier. Higher the value of MDCK cell, higher the cell permeability. Out of eighteen compounds sixteen designed compounds showed acceptable range of QPMDCK, fourteen compounds showed few number of stars viz; (IV), (X), (XI), (XII), (XIII), (XIV) showed zero stars, and (II), (VI), (VII), (VIII), (XV), (XVI), (XVII), (XVIII) showed one stars. Therefore these compounds showed most favorable results for anticonvulsant activity and the compounds that showed acceptable range of all descriptors can be assigned for further drug designing and also synthesize these compounds in laboratory. The most active compounds of our study were (IV), (X), (XI), (XII), (XIII), (XIV), (II), (VI), (VII), (VIII), (XV), (XVI), (XVII), (XVIII) because these designed compounds showed acceptable range of the descriptors. Drug likeness properties by molinspiration also showed good results showed in Table 5. On comparing the relative activity scores of diazepam with (I-XVIII) compounds utilizing the six drug classes, all the compounds showed similar bioactivity score as compared to the standard drug viz; Diazepam, especially in case of GPCR ligand, ions channel modulator, kinases inhibitor and nuclear receptor ligand.
Conclusion
A brief computational study was carried out over eighteen designed pyridazinone derivatives using various software programs with the goal of identifying potential lead molecules that efficiently bind to the human GABA receptors. Docking studies have helped us to know about the binding modes of hypothetical designed pyridazinone derivatives to determine their GABA facilitator activities. These investigations paved the way for the synthesis of selected compounds, which are more potent and selective GABA facilitator. Moreover, it was also proved from above discussion that geometry of receptor also plays a major role in defining drug action. Our ADME-T studies permit us to evaluate a set of anticonvulsant lead compounds and to assess the parameter that will be essential for further lead optimization efforts. The in silico evaluation confirmed that, the compounds had “druglike” properties but did not give information of sufficient value to discriminate between compounds. Succeeding lead optimization efforts are warranted to produce a fully optimized anticonvulsant drug candidate ready for preclinical development studies.
References
- Ekins S, Mestres J, Testa B (2007) In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br J Pharmacol 152: 9-20.
- Ballester GF, Carvajal AF, Gonzalez-Ros JM, Montiel AF (2011) Ionic Channels as Tartgets for Drug Design: A Review on Computational Methods. Pharmaceutics 3: 932-953.
- Moroy G, Martiny VY, Vayer P, Villoutreix BO, Miteva MA (2012) Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov Today 17: 44-55.
- Blume WT, Lüders HO, Mizrahi E, Tassinari C, van Emde Boas W, et al. (2001) Glossary of descriptive terminology for ictal semiology: report of the ILAE task force on classification and terminology. Epilepsia 42:1212-1218.
- Beyenburg S, Bauer J, Reuber M (2004) New drugs for the treatment of epilepsy: a practical approach. Postgrad Med J 80: 581-587.
- Edafiogho IO, Scott KR (1996) Burgers Medicinal Chemistry and Drug Discovery. John Wiley and Sons, New York, USA, Pp: 175.
- Cosford ND, Tehrani L, Roppe J, Schweiger E, Smith ND, et al. (2003) 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J Med Chem 46: 204-206.
- Jansen M, Dannhardt G (2003) Antagonists and agonists at the glycine site of the NMDA receptor for therapeutic interventions. Eur J Med Chem 38: 661-670.
- Czuczwar SJ, Patsalos PN (2001) The new generation of GABA enhancers. Potential in the treatment of epilepsy.CNS Drugs15: 339-350.
- Jannuzzi G, Cian P, Fattore C, Gatti G, Bartoli A, et al. (2000) A multicenter randomized controlled trial on the clinical impact of therapeutic drug monitoring in patients with newly diagnosed epilepsy. The Italian TDM Study Group in Epilepsy. Epilepsia 41: 222-230.
- Dam M, Bolwig T, Hertz M, Bajorec J, Lomax P, et al. (1984) Does seizure activity produce Purkinje cell loss? Epilepsia 25: 747-751.
- Allan RD, Johnston GAR, Kazlauskas R, Tran HW (1983) Synthesis of analogues of gamma-aminobutyric acid. Part 11. Unsaturated and saturated tetronic acid derivatives. J Chem Soc Perkin 2983-2985.
- Katritzky AR, Charles WR (1984) Pergamon press, UK, Pp: 2.
- Banerjee PS, Sharma PK (2011) New antiepileptic agents: structure-activity relationships. Med Chem Res 21: 1-18.
- Rubat C, Coudert P, Albuisson E, Basitde J, Couquelet J, et al. (1992) Synthesis of Mannich bases of arylidenepyridazinones as analgesic agents. J Pharm Sci 81: 1084-1087.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev 46: 3-26.

Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences